

The U.S. Federal Food and Drug Administration’s (FDA) origins as a federal consumer protection agency began with the passage of the 1906 Pure Food and Drugs Act. But its founding was not sufficient to prevent poor-quality food and medication from flooding the market. By 1938, in response to public outrage, Congress passed the Food, Drug, and Cosmetic Act, which granted the FDA increased authority in protecting U.S. consumers.
Early Success
Despite its shortcomings, the entirely taxpayer-funded FDA did once protect consumers. Rejecting the drug thalidomide is perhaps one of the most glorious examples of this protection. Thalidomide, used to treat morning sickness in pregnant women in the late 1950s, was approved by dozens of countries.
Dr. Frances Oldham Kelsey, one of only a handful of medical officers at the FDA at the time, was tasked with reviewing the thalidomide application. Despite constant pressure from the drug company, Dr. Kelsey refused to approve the application because she felt it lacked scientifically reliable evidence of the drug’s safety.

Approximately a year after thalidomide was marketed in other countries, researchers linked it to a debilitating birth defect that involved thousands of babies. Dr. Kelsey had been right in her stance. In 1962, President Kennedy conferred on Dr. Kelsey the President’s Award for Distinguished Federal Civilian Service.
AIDS and the Introduction of ‘User Fees’
In the 1960s and 1970s, there was a social movement in the United States and the developed world that challenged traditional norms of sexual behavior and relationships. Subsequently, in the 1980s, the AIDS epidemic ravaged the United States and the world. This triggered government agencies and pharmaceutical companies to put tremendous resources into developing new drug research to fight AIDS.
With sports stars and celebrities dying of AIDS-related complications, AIDS activists were outraged about long delays in getting experimental FDA-approved HIV drugs. They organized mass protests around the country, claiming that thousands of lives were lost each year due to delays in the FDA’s drug approval process. The protests and the massive lobbying in U.S. Congress proved to be effective.
In 1992, Congress passed the Prescription Drug User Fee Act (PDUFA), allowing drug makers to pay FDA “user fees” to speed up the drug application reviewing process.

The outcome was significant: The FDA’s new drug review staff increased by 77 percent, resulting in a 50 percent reduction in new-application review time and a 33 percent increase in medicines approved each year.
Essentially, the PDUFA of 1992 started the FDA’s transformation from providing oversight to providing review services for the drug industry—and from being solely taxpayer-funded to partially industry-funded. In other words, the FDA taking user fees from drug companies is like referees collecting lucrative fees from sports teams. The bigger the pharmaceutical companies are, the more user fees they pay to the FDA. Consequently, the more the FDA relies on these fees for its operation, the more influential these “Big Pharma” companies become.
A peer-reviewed article by The British Medical Journal (BMJ) quoted sociologist Donald W. Light of Rowan University in New Jersey, saying, “being largely funded by fees from the companies whose products it is charged to evaluate is a fundamental conflict of interest and a prime example of institutional corruption.” Light has spent decades studying drug regulations.
Fast-Track Approval for ‘Breakthrough Therapies’
From 1993 to 2016, user fees collected by the FDA from pharmaceutical companies have increased 30-fold—from around $29 million to $884 million. As the drug makers’ payment to the FDA became larger, so too did their influence over the FDA’s drug approval process.
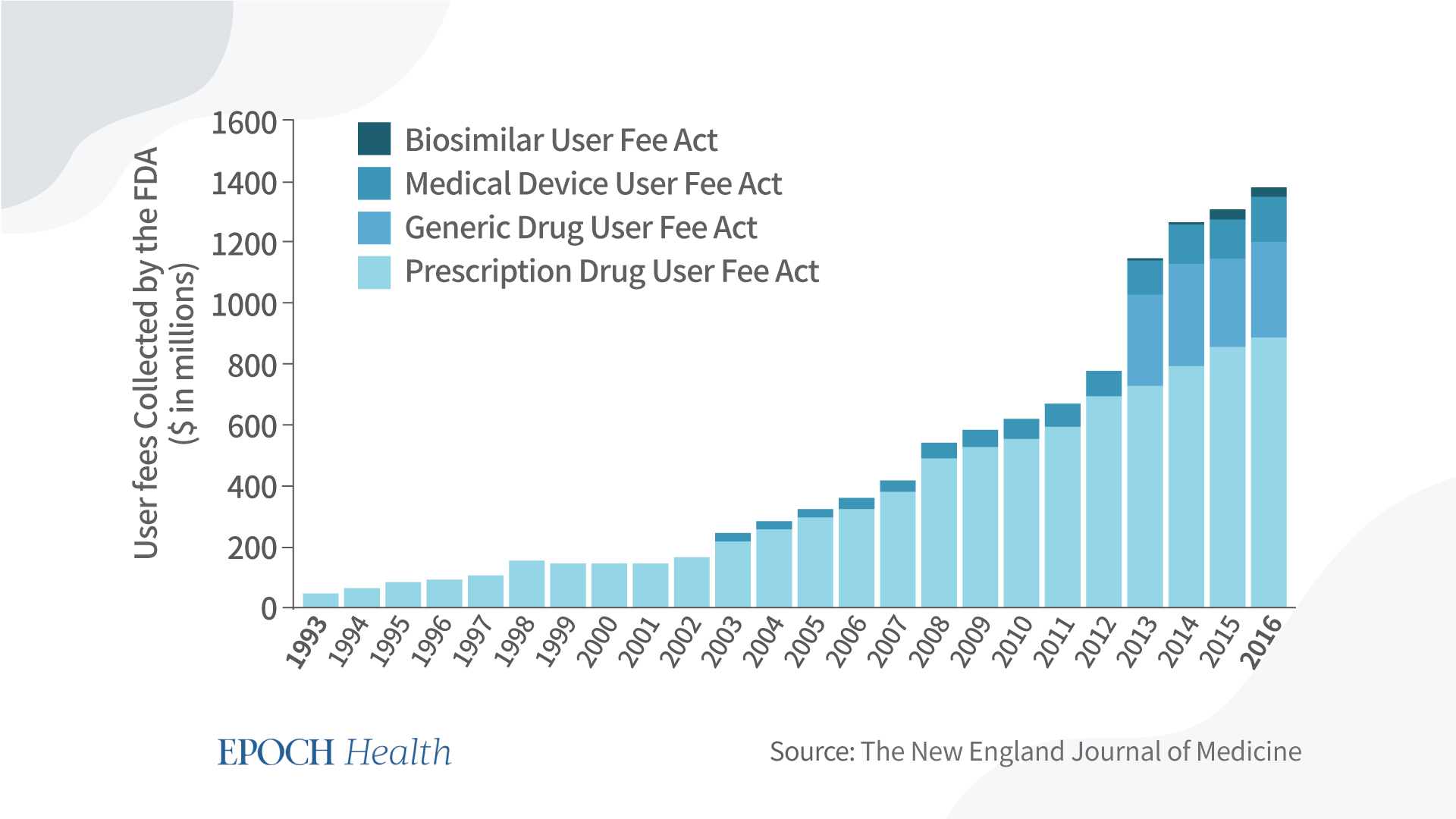
While the PDUFA was passed to speed up the process, the introduction of the “Advancing Breakthrough Therapies for Patients Act” of 2012 aimed to simplify the process. It allows certain drugs to be on a fast track toward approval, breaking the traditional drug approval process.
Two criteria qualifying a drug as “breakthrough therapy” are: It is intended to treat a serious life-threatening illness, and the preliminary clinical evidence indicates that the new drug may demonstrate improvement over existing therapies.
Who has the authority to grant the “breakthrough therapy” label to a new drug? The Secretary of Health and Human Services (HHS). Upon request by the drug developer, the secretary has 60 days to determine whether to label the drug a breakthrough therapy.
In addition, the Secretary of HHS must hire an independent entity to evaluate the FDA’s breakthrough therapy review process and report to Congress on an annual basis.

From July 2012 to December 2015, the FDA approved 40 out of the 384 breakthrough therapy requests.
However, “improvement over existing therapies” does not correlate with concerns about safety and toxicity. When the process is simplified under the “breakthrough therapy” umbrella, the drugs pursuing this fast-track approval often do not have clinical or preclinical studies presenting valid long-term or more comprehensive safety and toxicity data. Consequently, more drug recalls are inevitable.
What’s Next?
According to a study (pdf) published in The BMJ in June, “in 2020, 68% of drug approvals in the United States were through expedited pathways.” Accelerated approval processes have resulted in new drugs that were more likely to be withdrawn for safety reasons, more likely to carry a subsequent black-box warning, and more likely to have one or more dosage forms voluntarily discontinued by the manufacturer.
In 2021, the FDA gave fast-track approval to the Alzheimer’s drug Aduhelm, but it remains “unproven” by many experts, including FDA advisory committee members who were tasked to review the Aduhelm application.
Pfizer’s COVID-19 vaccine Comirnaty also gained full fast-track approval. And then throughout 2021 and 2022, there were numerous scientific reports, patient reports, and congressional hearings regarding the safety of this vaccine. How many people are harmed in the short term? Needless to say, we have no idea how many people will suffer long-term adverse effects.
The PDUFA was reauthorized by the U.S. Congress for the sixth time in October 2022. But when the PDUFA changed the relationship between the FDA and the drug companies, it also changed the FDA’s relationship with taxpayers. Can the FDA be trusted to protect taxpayers when a large chunk of its budget comes from Big Pharma? Will the cycle of more drugs being approved and more drugs being recalled come to an end?
History has told us repeatedly that people do foolish things that may result in harm to themselves in the name of the greater good. New legislation that can challenge the PDUFA status quo is certainly warranted. The question is: Who will have the guts to introduce it?
Views expressed in this article are the opinions of the author and do not necessarily reflect the views of The Epoch Times. Epoch Health welcomes professional discussion and friendly debate. To submit an opinion piece, please follow these guidelines and submit through our form here.